

For years, scientists in the field of molecular dynamics have been squeezed in their research. Traditional simulations that they have been using to track how atoms and molecules move through a system are limited in their accuracy. And recent efforts using machine learning have been tripped up by a lack of high-quality training data, which also hampers accuracy.
Moreover, quantum mechanical methods that promise greater accuracy can’t scale – due to compute demands and cost – to enable the modeling of biosystems with thousands of atoms.
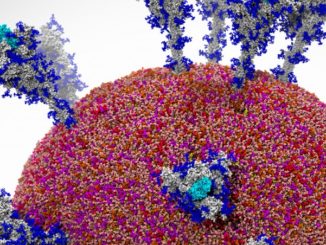
It has been a challenge in a scientific field that relies on accurate representations of the characteristics of molecules and atoms and predictions in how they work for everything from developing new therapeutic drugs and producing biofuels to creating medical biomaterials and recycling plastics.
However, scientists in Australia – with the help of the exascale-class “Frontier” supercomputer at Oak Ridge National Laboratory – spent four years creating software that can run a quantum molecular dynamics simulation that is 1,000 times faster and larger than what’s been done with previous models. The work was first announced in July and, last month, earned the scientists – along with researchers with chipmaker AMD and the Oak Ridge National Lab – the 2024 Gordon Bell Prize from the Association for Computing Machinery, which was presented at the annual Supercomputing Conference (SC24). We are circling back to tell you all about it.
In announcing the initiative during the summer, Giuseppe Barca, an associate professor at the University of Melbourne and the project’s leader, said the “breakthrough enables us to simulate drug behavior with an accuracy that rivals physical experiments. We can now observe not just the movement of a drug but also its quantum mechanical properties, such as bond breaking and formation, over time in a biological system. This is vital for assessing drug viability and designing new treatments.”
Barca also is head of R&D at QDX, a year-old Singapore-based startup that uses AI and quantum mechanics in its computational drug discovery platform and which also was part of the Barca-led project.
Unprecedented Granularity
The researchers said the simulation capabilities deliver details that let them see and understand these systems better than before, which will help drive drug discovery in an industry where, according to the University of Melbourne, more than 80 percent of disease-causing proteins can’t be treated with existing drugs and only 2 percent can work with the drugs now available.
Team members included Barca, five researchers from the School of Computing at the Australia National University, Jakub Kurzak, PMTS software system design engineer at AMD, and Dmytro Bykov, group lead in computing for chemistry and materials at Oak Ridge. They published their work in a 13-page paper titled Breaking the Million-Electron and 1 EFLOP/s Barriers: Biomolecular-Scale Ab Initio Molecular Dynamics Using MP2 Potentials.
Through its work, the group created a new technique beyond classical approaches and quantum mechanical by combining two methods, molecular fragmentation (the process of breaking down a large molecule into smaller fragments) and MP2 perturbation, a complex computational chemistry method that involves approximating the correlation energy of a molecule.
A Boost From Frontier
Using the supercomputer, the researchers were able to blow past the million-electron and 1 exaflops barriers for ab initio molecular dynamics, or AIMD, simulations with quantum accuracy. AIMD methods are used to combined molecular dynamics with quantum mechanics.
Frontier is a Hewlett Packard Enterprise Cray EX system that has reached 1.353 exaflops using almost 8.7 million cores and streaming multiprocessors across its CPUs and GPUs. The system, which is listed second in the latest Top500 list released last month of the world’s fastest supercomputers, is powered by custom “Trento” AMD Epyc CPUs that have been optimized for HPC and AI and AMD “Aldebaran” Instinct 250X GPU accelerators. It includes HPE’s “Rosetta” SIingshot-11 interconnect to tie them all together.
The researchers also ran calculations on “Perlmutter,” another HPE Cray EX supercomputer housed at the Lawrence Berkeley National Lab in California that ranks 19th on the Top500 list. It includes 4,864 nodes with a mix of AMD “Milan” Epyc 7763 processors and Nvidia A100 GPU accelerators as well as HPE’s Slingshot interconnect.
An Algorithm And Its Working Groups
The researchers in their paper said their “overarching algorithm” involved a multi-layer dynamic load distribution scheme using MPI, with a centralized super coordinator distributing polymers – large molecules comprising many smaller molecules that are chemically linked together – to worker groups from a centralized polymer priority queue.
“Worker groups are confined to a single node and can utilize any number of GPUs within the node,” they wrote. “There can also be several worker groups per node, each utilizing one or more GPUs. Each worker group has one rank per GPU with an additional local coordinator rank to enable dynamic load distribution within the group.”
Noting the limitations of previous methods – the lack of accuracy approaches that could scale and the “impractical” compute scaling that was needed for quantum-accurate ab initio – the researchers wrote that their work “enables a major leap of capabilities in computational chemistry and molecular dynamics, enabling the simulation of biomolecular-scale molecular systems with unprecedented accuracy. We overcome the limitations that previously confined quantum-accurate AIMD simulations to small systems. … These achievements are made possible through the exploitation of exascale supercomputing resources at unprecedented computational efficiency in computational chemistry simulations.”
In announcement in June of the work, Oak Ridge’s Bykov said that science like that done by the Australian researchers was “exactly why we built Frontier, to tackle larger, more complex problems facing society. By breaking the exascale barrier, these simulations push our computing capabilities into a brand new world of possibilities with unprecedented levels of sophistication and radically faster times to solution.”

Gordon Bell Prize Winners Leverage Machine Learning For Molecular Dynamics
For more than three decades, researchers have used a particular simulation method for molecular dynamics called Ab initio molecular dynamics, or AIMD, which has proven itself to be the method most accurate for analyzing how atoms and molecules move and interact over a fixed time period. In this time of …

Expanding The Search For A Range Of New Materials
Finding new functional materials for batteries and catalysts and lots of other uses is a major goal of researchers around the world. And the design and discovery of new materials often requires computer simulations running on the world’s fastest supercomputers using specialized software that can determine properties at the quantum …
Frontier: Step By Step, Over Decades, To Exascale
Any time you build anything with more than 60 million parts, it is going to be a headache. And if you have to create a space in a datacenter and build an exascale system with all of those parts, each of which is crucial, during a global pandemic, it gets …
Glad to see the Gordon Bell Prize awarded to this Ab Initio (from first principles) bioMolecular Dynamics (AIMD) computational research. If I understand, the approach is based on a post-Hartree-Fock (HF) Schrödinger equation wave-function-based (WF) second-order Møller-Plesset (MP2) perturbation method, that would normally scale as (N⁵) with problem size. The 2 million electron case was then tackled through parallelization over GPUs with a third-order many body expansion (MBE3) fragmentation approach that improves scaling to (N), and a resolution-of-the-identity (RI) approximation that replaces expensive four-center integrals with reusable three-center versions, followed by Verlet symplectic integration over asynchronous steps (Figs. 2, 4, and 7).
To me, the clincher comes with the results (in bold text) at the top-right of page 10: the computation of one time step takes “1.55 ZettaFLOPs on 9,400 nodes” of Frontier, and that takes 25.6 minutes of wall time (1,540 seconds) as the supermachine executes just North of 1 ExaFLOP per second … that’s what I call putting a supercomputer through its paces (bravo!)! And from this, and the needs of 1-km resolution climate modeling, it is becoming increasingly clear that we need to develop and qualify FP64 ZettaFlopping supermachines quick (let’s not rest on our ExaFlopping laurels!)!
One lingering question (for me, as a non-expert) is whether the specialized architecture of Anton 3 ( https://www.nextplatform.com/2023/12/04/the-bespoke-supercomputing-architecture-that-stood-test-of-time/ ) could help with such biologically-oriented AIMD computation, or if it is aimed at a slightly different conceptualization of the underlying processes, with a differing field of applicability? Either way, this work is very impressive imho, congratulations to the Barca (down under) team for winning this presitigous award, and let’s get ZettaFlopping soon, please!
Anton 3 is extremely hard-wired to doing electrostatics, whilst this work is much more about subtle quantum interactions – their big trick is to avoid having to do a calculation for every possible subset of four electrons, instead you do it for every subset of three electrons and can store the whole interactions for a thousand electrons in 40GB of GPU memory.
I’ve only ever used quantum chemistry tools as an input into a separate optimiser, modelling the ligand as a quantum object while doing the protein surrounding it with constraints much more like a ball-and-stick model, and where what we were trying to do is fit experimental X-ray diffraction data; I left the company fifteen years ago but it’s still there at the website in my link.