

In the history of computing, there has been an endless push and pull between the need for general-purpose versus fine-tuned custom systems and software.
While general purpose will, by nature, prevail on leadership-class HPC, the work done in meticulous world of ASIC design, system and software optimization filters into architectural thinking eventually. In the case of the present, ultra-specialization will likely come back around again for specific use cases in AI. One can argue it already has if the first wave of AI chip startups was any indication.
When it comes to special-purpose supercomputing, the go-to example is the Anton supercomputer architecture, a custom system dedicated to the task of solving (and dramatically speeding) complex problems in molecular dynamics with levels and speed and fidelity impossible even on top exascale supers.
Anton (and its founding father) were front of mind as it was formally recognized this year at the Supercomputing Conference (SC23) as a Test of Time Award winner. David Shaw, founder of research firm, D.E. Shaw, accepted the award and spoke at length about the evolution of the Anton system architecture and its algorithms, which have morphed to meet the times since the system’s 2008 unveiling.
Shaw represents a departure from tradition across the board. First, the Test of Time Awards have been largely centered on academic achievements. However, Shaw took a circuitous route to computational biology. After finishing his PhD at Stanford before teaching computer science at Columbia (while working on the NON-VON parallel system architecture) Shaw joined Morgan Stanley in the mid-1980s. He then founded a hedge fund, D.E. Shaw & Company, which focused initially on optimized trading algorithms before founding D.E. Shaw Research.
His work, whether for trading or grand-scale science challenges, emphasized speed and optimization on large parallel systems.
The prototype system, first described in an ACM paper in 2007, claimed that the massively parallel machine, “should be capable of executing millisecond-scale classical MD simulations of such biomolecular systems.”
The original paper also explained how the system, which was set to emerge in 2008 with “512 identical MD-specific ASICs that interact in a tightly coupled manner using a specialized high-speed communication network” could “dramatically accelerate those calculations that dominate the time required for a typical MD simulation.”
As it turned out, Anton 1 could do everything the D.E Shaw team outlined then. In the decade-plus of work on both the system architecture and application, in 2023, D.E. Shaw has six drugs in human clinical trials. Two were developed independently from concept to trial and four others were developed with Relay Therapeutics, which focuses on protein dynamics for identifying new drug candidates.
“Our long-term goal has always been to design new molecules that can serve as medications, which is something we’re finally doing, just in the last few years,” Shaw told the SC23 audience. He says that while his team is exploring the intersection of machine learning with the future of drug discovery, the supercomputing architecture piece has “been core all along and is still the most central.”
A central component to the Anton story over the years has been speed and scale—not just of the architecture (Anton 3 is 2X more powerful than the first generation) but of its ability to get the time scales of MD simulations down so far that complex interactions can be observed—well enough to design highly-targeted treatments for a range of treatments. Getting simulations down to the millisecond in 2008 was groundbreaking but Anton 3 can is in the 1-2 femtosecond range. As Shaw explained at SC23:
“In 2008, the fastest supercomputers of the time had simulated about 1/10th of a microsecond of time in a day. The longest that had ever been done was 10 microseconds and that lasted weeks or even months but was heroic computations. Many of the most important biological phenomena—the kind relevant to potential pharma design—all took place on scales of 10 microseconds so we were orders of magnitude away from where we needed to be.”
Those timescales meant that there wasn’t much to see other than molecules vibrating rather than the big changes that would allow for real discoveries.
Shaw demoed simulations that highlighted the value of ultra-fine timescales in drug discovery. In one, a protein shifts around, leaving gaps and pockets that the targeted medication can find and worm its way into. In another, he showed a hidden target which didn’t appear to have a binding entry point until simulations showed that there was an opportunity, something that wouldn’t have been possible if not for the ability to see activity that only lasted a very short time.
As it turns out, protein dynamics is exactly what the Anton machines do best. Protein folding, one of the key discoveries of our burgeoning century, opened doors for scientists beyond pharma. In fact, one of the most famous names in that arena, John Jumper, was working at D.E. Shaw Research during the early days of Anton development and went on to organize the AlphaFold project.
“New algorithms running on a conventional supercomputer would have been too slow. And a new supercomputing architecture running conventional algorithms would have been too slow.”
Shaw says that when the massively parallel Anton 1 machine emerged with its custom ASIC and special baked-in capabilities for particle interactions, it “allowed a dramatic increase in simulation length because of its speed—it was 100X faster than the fastest general purposes of the time and allowed continuous, millisecond-long simulations of proteins” which meant many new behaviors and interactions were observed for the first time ever.
Most of the chip area of the original machines (Anton 1 and 2) were dedicated to specialized math that honed in on the most computationally expensive parts of MD simulations. That meant there were (and still are) tradeoffs, including a lack of flexibility and programmability. It was “a very inflexible bunch of fast, stupid logic and nothing was programmable,” Shaw says. “We were brutal to the people who were designing that embedded software and put high priority on having it run fast.”
The data flow nature of the Anton systems meant that data went right to where it was needed, it didn’t stop along the way or pop to global memory. There’s plenty of memory on the systems but it’s distributed across the chip, which meant to high bandwidth and low latency. “At the interchip level, we had some app-specific ways of minimizing latency and the overall throughput but overall, we always had the luxury of doing that because we knew what algorithms we were trying to speed.
By the way, if this architectural discovery sounds familiar, it is of course happening among all the AI chip players who, in some ways, also have the luxury of a defined workload to optimize around.
By 2013 with the introduction of Anton 2, teams reported Anton 2 was an order of magnitude faster than the original with support for 15X as many atoms. They were able to add better flexibility and programmability and support for more accurate physical models with some new algorithms. The capacity, or total number of atoms, was big deal for potential discoveries but it meant more data movement between chips and further refinement to communication strategies.
With Anton 3 last year, D.E. Shaw pushed its 512-node machine into public view, showcasing its ability to simulate biological systems at unprecedented scale—in the ballpark of millions of atoms. “There were a number of architectural changes in this machine due to changes in underlying technologies, including different rates of in advancement in processing versus communication parameters” but it meant a new world of discoveries, including those that led to the drugs in clinical trials now.
The following slide highlights the improvements compared to traditional HPC architectural elements (GPU/CPU).
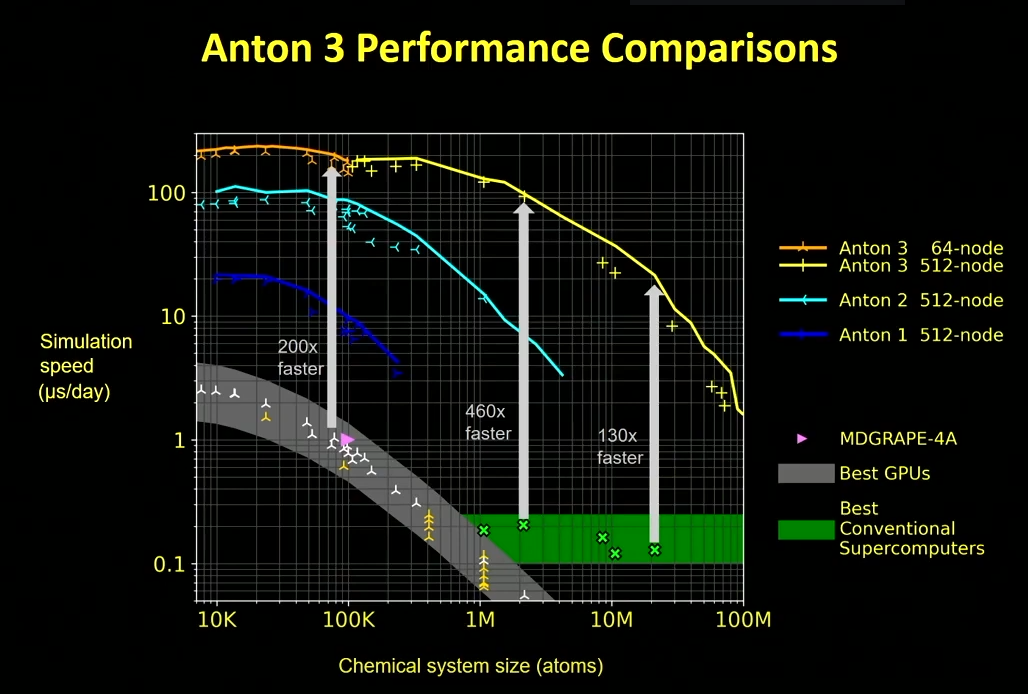
The x axis is size of the biological system, the simulation speed is on the y axis (microseconds per day). As expected, the curves show what we see elsewhere: as you go up in system size performance goes down (simulating more interactions).
Anton 3 had some of the largest jumps architecturally, including refactoring the ASIC layout to minimize increasingly expensive cross-chip communication by moving to a tile-based architecture that combines sub-tiles for both the “hardwired” particle interaction pipelines and also programmable and more flexible processing units.
Designers also added specialized “bond calculators” or modules that sped up some of the slower parts of working with different bonded atoms and the interactions they propagate. “We could save area, energy, and get speed with this chunk of hardware to take the load off other parts of the chip,” Shaw explains. The team also worked on some novel data compression techniques but ultimately, communication proved the bottleneck. “We learned about our calculations and looked in detail at the underlying physics to find redundancies and opportunities but we have the luxury of looking at just one application.”
“Historically, it’s been hard to have special purpose machines compete with general purpose supercomputers. It was great that Anton got 100X for this application—exciting to us. But the part that relates most to our long-term goal was more to do with the underlying science, learning about molecular systems and curing people. That’s what we wanted to do.”


The Huge Payoff Of Extreme Co-Design In Molecular Dynamics
When money is really no object, and the budget negotiations involve taking a small slice of your personal net worth of $7.5 billion out of one pocket and putting it into another, and you have the technical chops to understand the complexities of molecular dynamics, and have a personal mission …
The Evolution of NAMD: A Scalability Story from Single-Core to GPU Boosted
On today’s episode of “The Interview” with The Next Platform, we take a look at the evolution of the NAMD molecular dynamics and how the introduction of GPU computing upended performance expectations and set the stage for new metrics now that the Volta GPU architecture will be available on large …
A MapReduce Accelerator to Tackle Molecular Dynamics
Novel architectures are born out of necessity and for some applications, including molecular dynamics, there have been endless attempts to push parallel performance. In this area, there are already numerous approaches to acceleration. At the highest end is the custom ASIC-driven Anton machine from D.E. Shaw, which is the fastest …
Fantastic article (IMHO)! It’s great to read about this “priceless” “fire-breathing monster for molecular dynamics simulations” (from first link, “Anton”, with many diagrams and photos). The second link, “NON-VON”, from 1982, is also superb; on page 6 we find:
“In the NON-VON primary processing Subsystem (PPS) […] a large number of very simple […] processing elements (PE’s) are, in effect, distributed throughout the memory.”
Right on! That’s near-memory (or in-memory) dataflow-style compute right there (as you certainly noted: “The data flow nature of the Anton systems […]”)!
Now, if this arch could be merged with that of AI’s fine-grained reconfigurable dataflow accelerators (usual suspects; not the chunky ones), it would make a real AI/MD multipurpose winner (I think)!
I didn’t include this in the piece but I wonder if even this MD powerhouse will be less effective than GenAI for drug discovery. I suspect you might know a thing or two about this there given your bio background there in MD. What do you think?
Currently there is basically no public data (databases) for the conformational landscape of proteins, so no training data to use AI to do what they’re doing with MD. It’s not impossible to create it, e.g. with active learning using MD, but I’d say this is not going to happen on the short-term. 5 years from now, maybe.
Alphafold-like models are already having big difficulties with the lack of training data, see RosettaFoldNA with nucleic acid protein complexes…
Great, useful answer. Thanks for the insight.
I’m pretty much with Fabio on this, in that ML-oriented AI approaches require a lot of training data which may not be readily available for drug discovery applications at present (unlike for LLMs). Dr. Matysiak (former UMCP colleague: https://bioe.umd.edu/clark/faculty/169/Silvina-Matysiak ) is a folding expert who could likely provide a more nuanced answer on some of this.
I’ll note also the UMCP’s recent DOE INCITE award (Nov. 29) of 100’s of thousands of node-hours on Aurora, Frontier, and Polaris, to advance AI/ML training and application on HPC machines, including healthcare and physician support (if I read well: https://www.umiacs.umd.edu/about-us/news/umd-team-wins-doe-award-advance-ai-using-supercomputers ). Drug discovery might be a component of this I guess, if there is sufficient faculty interest and availability.
Meanwhile, I would expect that some AI approaches that are not particularly rooted in ML (or in the way that ML is currently applied) could find uses in drug discovery. For example, AI-oriented cognitive modeling and optimization strategies could surely be useful here, especially in combination with MD (IMHO).
From the angle of computational processes and architecture, in AI we have many artificial neurons acting in parallel and interacting through synapses (linear vector biasing, linear matrix-vector multiplication, nonlinear activation indepedent of other neurons), and in MD we have many atoms or molecules acting partly in parallel (momentum) and partly in interaction with others (eg. distance-dependent nonlinear Lennard-Jones potential) which is slightly more complex (symplectic integration could be a must?). So I think an arch designed for MD could work well in AI but not necessarily the reverse. Also, AI can benefit from lower precision but I’m not sure if MD can too (maybe mixed-precision?).
In any instance, if genAI can somehow speed up drug discovery, then so much the better I think (IMHO)!
This is a super-fast moving field with enormous stakes and outsized competition between AIslingers — a wild-wild west exploration of new drug discovery frontiers, with associated fevers, snake oils, and journalistic opportunities (so we, readers, can keep up, without being trampled by the stampede!). In this rodeo atmosphere, one can expect wild claims and counterclaims by competitng vendors (look! gold!?), and for academia and boffins in gov labs to offer a more objective and modulated perspective.
For example, from Facebook folks (2021: https://www.pnas.org/doi/10.1073/pnas.2016239118 ):
“Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences” (wowee!)
and, from Meta folks (2022: https://www.biorxiv.org/content/10.1101/2022.07.20.500902v1.full ):
“Language models of protein sequences at the scale of evolution enable accurate structure prediction” (yippee!)
But, from 58 highly sedated academics (Sept. 2023, paywalled: https://www.nature.com/articles/s41573-023-00774-7 ), on “Artificial intelligence for natural product drug discovery” (a review paper), we find in the abstract:
“We also discuss how to address key challenges […] such as the need for high-quality datasets to train deep learning algorithms and […] algorithm validation”
Geez Lewise, what party poopers! They probably just forgot to read the industry papers! You snooze, you loze! (eh-eh-eh!)
Interpolating suggests that there is great potential but kinks still need ironing out …
It would be interesting to understand where you are today given the recent advances in AlphaFold and related protein design tooling coming from DeepMind. BTW are they now your competitors in drug design going forward?