

BioTeam, the famous HPC consulting practice, is using the quad-socket large memory nodes at the Texas Advanced Computing Center to speed alignment and HMM (Hidden Markov Model) inference workflows to find biological “synonyms” in biological databases like GenBank. This work gives hands-on bench scientists a collection of HMMs that reference a single coronavirus gene or protein so they can create complete, curated datasets of genes or proteins to use in their experiments to find vaccines and novel ways to treat diseases.
Brian Osborne, director of science at BioTeam, describes the TACC large memory node as a “superserver” for their bioinformatics workflows that will help scientists target coronavirus (specifically SARS-CoV-2 as it is known, often shortened to CoV-2) vulnerabilities, and this virus is the one, of course, that causes the COVID-19 disease that is infecting the world.
“I appreciate all the cores and memory in the TACC large memory nodes because whether I run one or four scripted workflows simultaneously doesn’t seem to make any difference,” says Osborne. “In that sense, it’s a great bioinformatics machine, I can’t seem to slow it down.”
Osborne explains the importance of BioTeam’s work as follows: “We use HMMs to turn alignments into a statistical representation. HMMs are particularly good at finding subtle similarities. The transmembrane domain is one example as the HMM is an extremely sensitive detector and can find sequence features that sequence similarity searches can’t.” In CoV-2 research, for example, transmembrane domains are involved in the formation of the spike protein that the virus uses to infect cells. Scientists can potentially exploit vulnerabilities in these different protein domains to fight the disease. The BioTeam research is entirely volunteer, and all work is open source on GitHub.
“Sequence database homology searching is one of the most important applications in computational molecular biology,” Sean Eddy wrote in his paper Accelerated Profile HMM Searches. “Genome sequences are being acquired rapidly for an ever-widening array of species. To make maximal use of sequence data, we want to maximize the power of computational sequence comparison tools to detect remote homologies between these sequences, to learn clues to their functions and evolutionary histories.”
Tying sequences to function is core to bioinformatics research. Along with sequence alignment, homology searches and HMMs are basic tools for these tasks. For this reason, the BioTeam computational workflow uses the well-known and respected, open source HMMER (Biological sequence analysis using profile hidden Markov models) package by Sean Eddy and the HMMER development team to create curated datasets that other bench biologists find useful. More precisely, the BioTeam code will:
- Download the tens of thousands of COV genomes from GenBank using taxonomy identifiers.
- Perform the lexical and semantic work to classify sequences via synonyms. This created a dictionary of co-terms that reference a single gene or protein.
- Create a collection of each COV protein and gene, taken from all the genomes.
- Create an alignment for each protein and nucleotide collection.
- Create a protein and nucleotide HMM based on each alignment.
- Use the nucleotide HMM to fully annotate all the genomes, which are mostly partially annotated, and search sequence databases.
Computational Challenges
Unlike neural networks and deep learning, HMM inference is challenging to implement on massively parallel SIMD architectures. For this reason, CPUs are the workhorse computational platform.
According to the HMMER documentation: “The latest version is highly optimized for performance on modern multi-core CPUs with SSE capabilities.” Homology detection on CPUs, which uses the MSV filter, “is very, very fast.” So fast that the User Guide notes that it is often input-bound rather than CPU bound.
For those interested in the computational details, the latest version of hmmsearch in HMMER 3.3.1 utilizes heuristic-pipeline which consists of MSV/SSV (Multiple/Single ungapped Segment Viterbi) stage, P7 Viterbi stage and the Forward scoring stage to accelerate homology detection. This pipeline is written as a very tight and efficient piece of assembly language code that makes use of the per core vector capabilities of modern CPUs.
Large memory computational nodes speed HMMER workloads because all the data can be kept in memory, thus bypassing the IO bottleneck. Osborne notes that early in the project, April 2020, there were 900 Coronavirus sequences in GenBank. Currently there are over 30,000 sequences of which most are partially annotated with the annotation being focused on the viral spike sequence and other structural proteins being targeted for vaccines.
The rapid growth in unannotated sequence data is a trend reflected throughout the bioinformatics community. Meng and Ji noted in their article Modern Computational Techniques for the HMMER Sequence Analysis, “The tendency is likely only to be reinforced by new generation sequencers, for example, Illumina HiSeq 2500 generating up to 120 Gb of data in 17 hours per run. Data in itself is almost useless until it is analyzed and correctly interpreted. The draft of the human genome has given us a genetic list of what is necessary for building a human: approximately 35,000 [coding and non-coding] genes.”
TACC is home to Frontera, the largest academic supercomputer in the world, which was specifically designed to support a range of intense academic workloads, from large memory to floating point intensive.
Frontera hosts 16 large memory nodes which require special access via the NVDIMM queue. The naming of the queue reflects the fact that these nodes contain 2.1 TB of Intel Optane persistent memory although the TACC large memory nodes also provide 112 Intel Xeon SP-8280M Platinum cores (28 cores per socket in a quad socket configuration) as opposed to the 56 Intel Xeon SP-8280 Platinum cores (28 cores per socket in a dual socket configuration). The large memory capacity and core count of these nodes provide Frontera users with extraordinary research tool and glimpse into the future
Once granted an account on Frontera, BioTeam then requested and were granted access to the special NVDIMM runtime queue. This runtime queue ensures that jobs that require the special capabilities of the large memory nodes will run only on the large memory nodes.
BioTeam was able to immediately able to start its investigation – no program modifications were required – because TACC configured the Intel Optane persistent memory to run in memory mode. Further the TACC systems team did not have to concern themselves with the two-tier nature of the Optane persistent memory and 384 GB DDR4 memory operations (shown below) as even the operating system on the Frontera large memory nodes were unaware of the Optane memory.
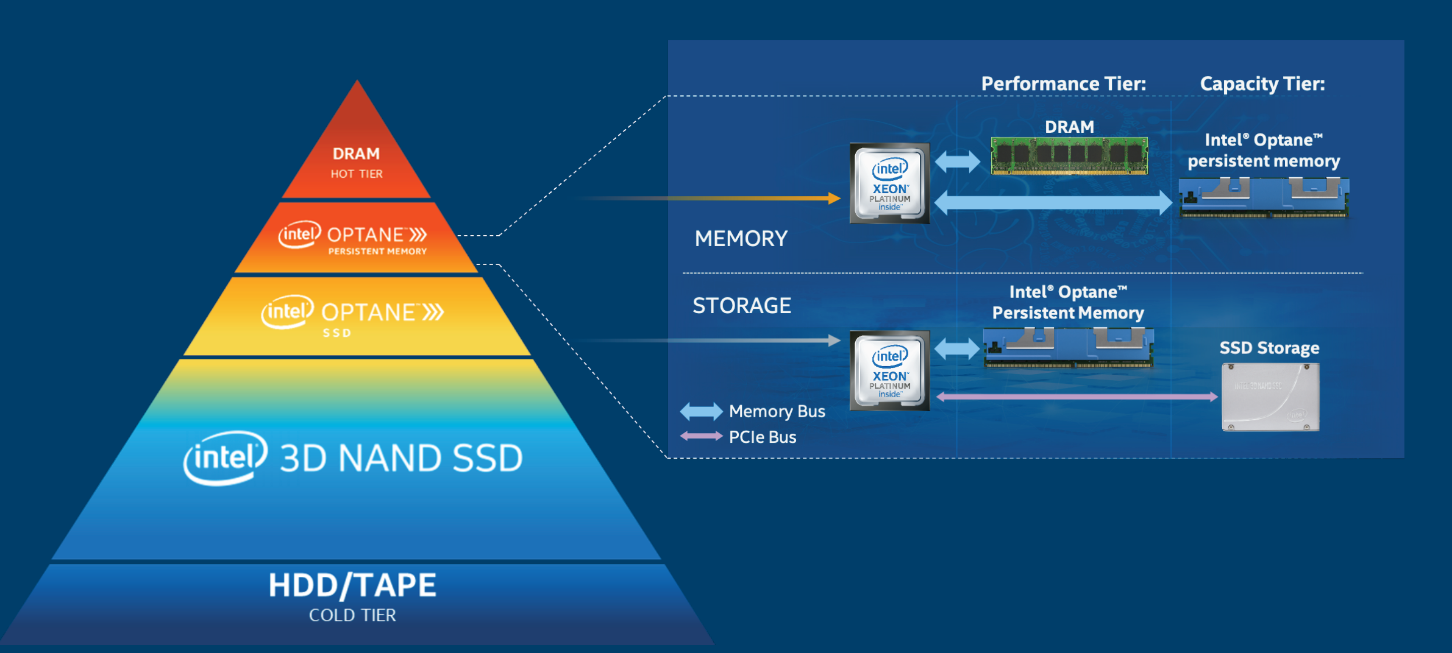
The reason is that in memory mode, the DRAM acts as a cache for the most frequently-accessed data while Optane persistent memory provides the large memory capacity. Specifically, the CPU memory controller will first attempt to retrieve data from the DRAM cache. When the data is present it will return the request from the DRAM cache, similar to the way DRAM access works today. When the data is not present, the request will be sent to Optane PMem. The request is returned to the CPU and in parallel is sent to the DRAM cache. Thus, the TACC large memory nodes simply look like they contain 2.1 TB of physical memory. No operating system intervention is required.
BioTeam exploited the 112 processor cores and 2.1 TB memory space by running up to four workflows at the same time – including alignment and HMM inference operations. This meant that BioTeam could generate results quickly, an important consideration when investigating a virus currently causing a global pandemic.
In particular Osborne notes, “Alignments are extremely fast on the Frontera large memory nodes. It’s kind of like running on a superserver with hundreds of cores.”
The speed of the HMMER alignments on the TACC large memory nodes reflects all the design effort that went into delivering the performant large capacity 2.1 TB hierarchical memory system as the HMMER User Guide notes: “The HMMER ensemble alignment algorithms (the HMM Forward and Backward algorithms) are expensive in memory.”
Creating Curated Datasets To Fight Disease
Tying everything together, Osborne notes that non-structural proteins (for example NSP1, NSP2) are usually not comprehensively annotated in GenBank entries. In the absence of annotation, BioTeam used HMM homology searches and the synonyms discussed at the beginning of this article to identify genes in sequence databases even in the absence of annotations. This ability to “pluck out” genes for proteins is used to identify interesting high-fidelity homology matches outside of the viral world. The end result is the creation of curated datasets that scientists can use in their studies about the all-important sequence to structure/function relationship. If you understand these relationships, then you can target vulnerabilities that can be exploited to create a vaccine or find a cure for a disease. Contact BioTeam to learn more and get access to these curated datasets, or run the code yourself.
Not just limited to Covid-19 research, which is understandably of extreme interest right now, the BioTeam work is also applicable to other viruses such as SARS (Severe Acute Respiratory Syndrome), MERS (Middle East Respiratory Syndrome), and many others.
Rob Farber is a global technology consultant and author with an extensive background in HPC and in developing machine learning technology that he applies at national labs and commercial organizations. Rob can be reached at info@techenablement.com.

Attacking The Novel Coronavirus With Supercomputing Cycles
Dan Stanzione has a lot of compute power at his fingertips. As executive director of the Texas Advanced Computing Center (TACC) in Austin, Stanzione is in charge of a number of supercomputer, including “Frontera,” a Dell EMC machine powered by Xeon SP Platinum processors from Intel that was deployed last …

If NSF Snoozes, Then TACC’s “Horizon” Supercomputer Loses
It is a tumultuous time for any agency in the US government or any company or organization that depends on the US government for a sizable portion of its funding or revenue. The National Science Foundation is front and center on proposed cuts in spending by the Trump administration, and …

Stampede3: A Smaller HPC System That Will Get More Work Done
All of the major HPC centers of the world, whether they are funded by straight science or nuclear weapons management, have enough need and enough money to have two classes of supercomputers. They have a so-called capability-class machine, which stretches the performance envelope, and a capacity-class machine, which plays an …
Be the first to comment