



DNA sequencing has been driving unprecedented discoveries in the life sciences since the emergence of next-generation sequencing (NGS) technologies ten years ago. With the cost of sequencing a human genome now falling below $1,000, the reality of personalized medicine is coming into focus and DNA sequencing is at the heart of it all. However, every sequenced genome is the product of an extensive computational process that transforms hundreds of gigabytes of raw sequencer output into aligned genomes, and the sequencing industry has been demanding increasingly larger compute and storage resources to keep up.
Large sequencing core facilities are now facing many of the scale-out issues that high-performance computing (HPC) has been addressing for years, and the smart sequencing centers have been quick to realize that hiring people with backgrounds in HPC operations positions them ahead of the curve on managing large-scale computing infrastructure. And given the famous fact that DNA sequencing technology is outpacing Moore’s law, this scale-out will have to accelerate—that is, each new generation of sequencer will require more, not simply faster, compute and storage.
Despite the growing size of compute and storage clusters in sequencing shops, the field of sequencing-driven computational biology is not actually doing HPC yet. In fact, it is unlikely that the important scientific questions to be answered by bioinformatics in the near future will require supercomputers. Rather, the computational demands of DNA sequencing can be satisfied more economically by using lab-scale and enterprise-oriented technologies.
To elaborate, it is meaningful to examine the major touch points between bioinformatics and HPC cyberinfrastructure: (1) the computational workload coming from DNA sequencing, (2) the critical performance parameters that drive effective system architectures for the given workloads, and (3) the algorithms and applications being used to advance the field.
Computational Workload
Data parallelism and statistical methods lie at the core of all DNA sequencing workloads because the physical process of decoding DNA is error-prone. These errors and uncertainties of errors are effectively mitigated by sequencing the same pieces of DNA many times over and calculating quality scores—an indication how likely any given data point may be an error—which propagate through virtually every calculation performed across the entire genomic dataset. At a very high level, the general process working with sequenced DNA can be broken down into three steps each with their own storage and compute requirements.
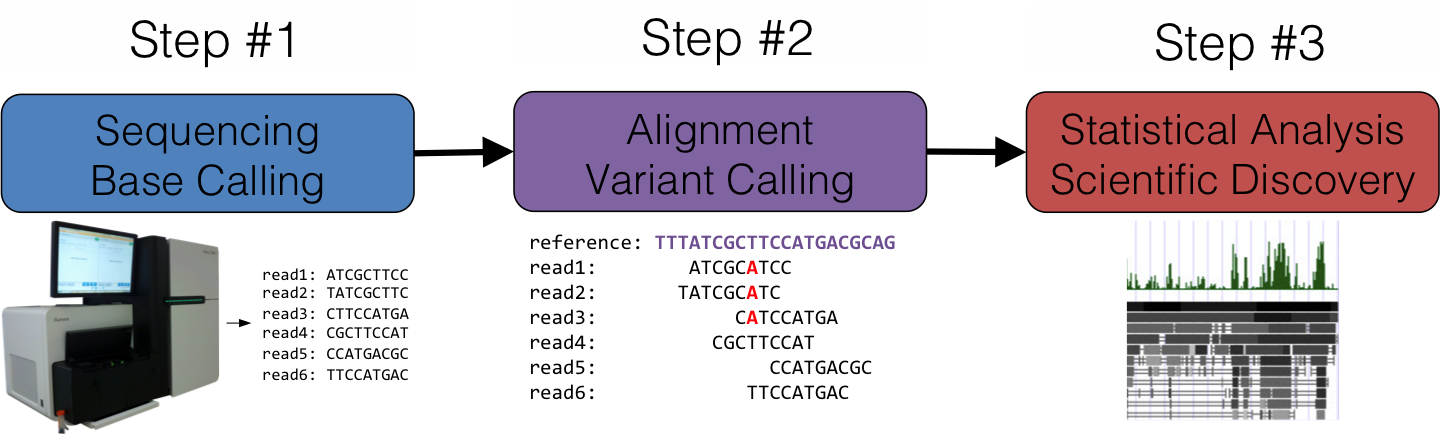
Step #1 involves running the physical sequencer instrument (“sequencing”) and converting the raw output from the optical or electronic sensors into millions of short reads comprised of As, Ts, Gs, and Cs (“base calling”). All computing for this step is carried out on the sequencing instrument itself, placing a minimal burden on external compute infrastructure. Furthermore, the output data is calculated in near real-time and continuously copied to a network file system, resulting in network data movement that averages around a dozen megabits/sec over the course of a days-long sequencer run.
Step #2 maps the millions of short reads generated by Step #1 to a reference genome (“alignment”) to determine how all those reads fit together. Once those millions of reads are pieced together, the parts of the aligned sequence that differ from the reference genome are called out (“variant calling”) and saved as a more compact dataset. This step can double or triple the size of the dataset that came out of Step #1, require up to ten times that space for scratch space, and consume hundreds or thousands of core hours.
Step #3 takes the called variants and tries to correlate those differences with scientifically or medically meaningful phenomena. This may involve cross referencing the called variants with databases of known variants, trying to find undocumented variants shared by a common group of patients who carry the same medical condition, or any number of other higher-order analyses.
The lion’s share of computing resources are consumed during Step #2, which often takes the form of a dozen or more applications that are strung together into a pipeline to progressively refine and error-correct the alignment and called variants. Most sequencing shops will carry the sequencing through the end of Step #2 before handing the data back to the biologists since these pipelines are fairly standardized and follow a common conceptual procedure. Although researchers tend to have their own choices of specific applications and options for variant calling pipelines, Step #2 generally varies very little between sequencer runs at any given sequencing shop and the computational demands are driven by high-throughput, not high-performance, requirements.
Rather, the innovation and scientific discovery happens at Stage #3, and unlike the compute-intensive Step #2, Step #3’s analyses are most commonly performed on lab-scale computing infrastructure. The tools and analyses are often of custom design and implemented with relatively low-performance but highly usable languages like R, Python, and Perl, and many of the analyses that resemble SQL-like operations benefit from storing the genomic data in relational databases. While Python’s utility as an HPC language is steadily growing, R is beginning to show greater support for parallelism, and SQL backed by HPC has shown great promise in bioinformatics, the effective integration of these tools with traditional HPC infrastructure is still in its infancy.
It is simply far more common to see many copies of the same R or Python script analyzing different subsets of a genomic dataset than it is to see a single parallelized application analyzing a whole genome across multiple nodes. This is a result of two major factors:
- There is still tremendous discovery to be made using workstation-scale analyses of genomic data, so the investment in software engineering is not worth the returns
- Sequencing is still too expensive to assemble large enough datasets to drive statistical analyses of variants that would require tightly coupled, multi-node calculations
Neither of these two factors are fundamentally limiting, and it is very likely that more complex analyses that find complex relationships across whole genomes or large sample sets will become more common in the coming years. It is likely that these calculations, which can involve solving very large sparse matrices, will turn to true HPC methodologies when the underlying science becomes rate limited by the speed of R and Python. The beginnings of this evolution is already evident with bioinformatics showing up on GPUs, Xeon Phis, FPGAs, other specialized ASICs, but the reality is that these accelerated applications are almost exclusively demonstrating proofs of concept, addressing only a small subset of problems, or solving trivially parallel problems.
Network Topology
The majority of the computational burden that lies downstream of DNA sequencing resides in the routine alignment and variant calling processes, which tend to be very data-parallel. Like applications in HPC, this variant calling pipeline puts a large stress on networking as a result of the large volumes of data that have to be moved between network-attached storage and the nodes performing the computations. Unlike conventional HPC, though, virtually none of this high-bandwidth network traffic moves between compute nodes; rather, the most critical data path lies between the compute fabric and the storage interconnect. As such, optimizing the cluster topology for a large amount of ingress and exit bandwidth rather than fabric bisection bandwidth is key.
Consider a typical compute-oriented system like NERSC’s Cray XE6, Hopper, which has 6,384 compute nodes connected in a 3D torus. While it has a tremendous bisection bandwidth within the compute fabric (over 3.8 TB/sec each way), data that has to leave this fabric to go to disk is funneled through 56 XIO nodes connecting out to Hopper’s scratch file system. This configuration provides a much more modest cross-sectional bandwidth between compute and storage of 224 GB/sec so that, at peak load, each compute node can only read and write data to storage at about 36 MB/sec.
To put this 36 MB/sec into perspective, consider the following I/O profile from a job that was sorting around 630 gigabases of DNA:

For this job, a single node was reading at an average rate of 930 MB/sec over 5.5 hours, meaning that only 247 of Hopper’s 6,384 compute nodes would be able to run this sort of job before fully saturating the I/O nodes. Granted, this read rate of 930 MB/sec per node is at the extreme end of what jobs in bioinformatics (or otherwise) will demand, but this sort of load would not be unimaginable on a sequencing analysis cluster if multiple DNA sequencers finished sequencing at the same time.
Thus, sequencing operations require clusters with decidedly data-oriented designs. Data-intensive clusters like Gordon at SDSC represent a significant step in this direction; like Hopper, it has a 3D torus interconnect that provides a modest 384 GB/sec bisection bandwidth across its compute fabric[1]. However, the storage subsystem is connected to this compute fabric at every point on the 3D torus, providing two major benefits for Gordon’s I/O capabilities:
- Every compute node shares an InfiniBand switch with an I/O node that routes to the storage fabric. This guarantees a single-hop data path from compute to storage that never has to touch the torus fabric[2].
- The aggregate ratio of compute to I/O node is an impressive 16:1 (cf. Blue Waters’ 56:1, Hopper’s 114:1, or HECToR’s 234:1).
These two factors provide a cross-sectional bandwidth of 160 GB/sec between compute and storage fabrics despite the storage fabric being based on 20 Gb/s Ethernet instead of 32 Gb/s QDR InfiniBand. This configuration affords each compute node 156 MB/sec at full I/O load, or 4X the amount available to a system like Hopper.
This is not to say that sequencing clusters are best served by torus interconnects; the benefits of rich cross-sectional bandwidth between compute and storage apply to other topologies all the same. In fact, the most cost-effective network topology for a sequencing cluster would forego bisection bandwidth entirely (i.e., no discrete high-performance compute fabric) and instead consist of smaller compute clusters that each have their own I/O node that routes directly to the storage fabric. This configuration minimizes the cost of providing high-bandwidth connectivity between compute and storage by eliminating unused additional networking.
IO Workload
The most IO-intensive operations in HPC applications are those where the state of the entire simulated system (for example, the x,y, and z position of every atom making up a virus capsid) needs to be checkpointed, or saved to disk. Because every job node stores its portion of the simulation in DRAM, every node must effectively flush the contents of its memory to a file system at the same time to capture a fully consistent checkpoint. High-performance parallel file systems like Lustre are specifically designed to cope with this massively parallel I/O so that one very large simulation can construct a single checkpoint file on a single file system. But checkpointing remains one of the most challenging scalability issues in HPC and is a focus of ongoing R&D effort.
The I/O profile of DNA sequence alignment and variant calling pipelines do not demand the same strict concurrency because of their lack of tight coupling. While the alignment of hundreds of human genomes can still impose huge aggregate bandwidth requirements (as in the case of the aforementioned 900 MB/sec/node sorting operation), each node does its share of the work independently of all others. This simplifies the file system requirements on sequencing clusters in two ways:
- A single logical operation (e.g., aligning a hundred genomes) does not have to stall (wait at a parallel barrier) until all nodes have written their intermediate results to disk. As soon as a single node has checkpointed its data, it can continue on with its calculation while other nodes are still writing. This mitigates the overall loss of compute efficiency when storage bandwidth is oversubscribed because I/O bandwidth becomes a problem of capacity, not capability.
- Data does not have to be stored on a single file system because genomic data can be split up rather arbitrarily. Since most bioinformatics applications will happily operate on sharded data, it is often easier to store a genomic dataset across several lower-performance network file systems than cope with the costs and complexities of scaling out a single large parallel file system.
The tradeoff for DNA sequencing is that their I/O workloads tend to demand many more I/O operations per second (IOPS) than HPC applications as a result of their data being distributed across many files. Parallel file systems are notoriously ill-suited for the metadata-heavy workloads associated with operating on many smaller files, so overall application performance can actually benefit from using multiple network file systems instead of a single parallel file system:
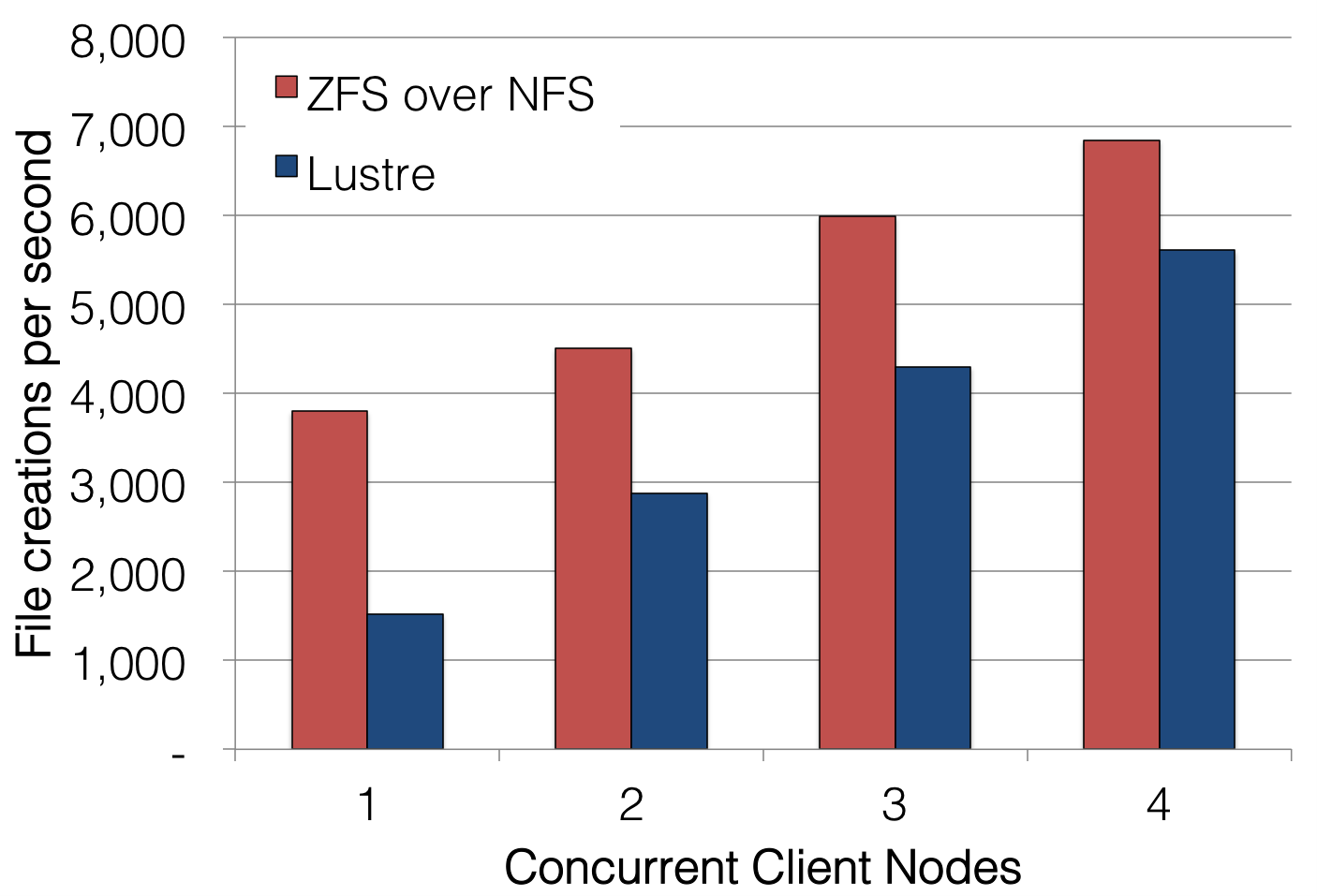
Even at low concurrency, a high-performance NFS server can out-perform a parallel file system on metadata-intensive tasks such as file creation. At higher concurrency, more NFS servers can be added to scale out this metadata performance linearly since DNA sequence reads, aligned reads, and called variants are all easily sharded into multiple files across multiple file systems. Parallel file systems, on the other hand, show much more modest scaling even as additional metadata servers are added to the file system, so the benefits of sharding genomic data cannot be efficiently realized on them.
Data Lifecycle
The lifecycle of data, or the processes by which data is generated, moved, refined, and removed from the storage subsystem, is also a critical consideration when designing data-intensive computing systems because it can dictate the optimal balance of performance, capacity, and policy. Typical HPC applications and sequencing pipelines actually share a common data lifecycle in a broad sense:
| Step in Lifecycle | HPC Application (e.g., |
Sequencing Pipeline (e.g., whole-genome study of a clinical trial) |
| 1. Read an input dataset | Reading checkpoint file from previous job | Ingesting raw output from DNA sequencer |
| 2. Periodically dump data during calculation | Dump configuration of entire system for ex situ analysis | Save successively more refined sequence alignments |
| 3. Write a large final output | Dump final configuration of system as well as additional metrics for a full restart | Save a fully finished and aligned genome |
| 4. Delete intermediate scratch data | ||
| 5. Move data to lower-performance, higher-capacity storage for analysis | ||
| 6. Reduce data to only valuable components and move to offline storage | Downsample data and save to tape | Upload reads and quality scores to Amazon S3 |
However, the critical difference between HPC and sequencing workload lifecycles is the strong coupling within distributed HPC applications. This coupling results in bursts of high-intensity parallel I/O during lifecycle Steps #1 through #3, at which time a significant fraction of the cluster’s high-performance scratch file system can be “hot.” By comparison, sequencing analysis pipelines’ executions are very loosely coupled, and it is more common for them to cause small hot spots across the file system that move as nodes process their own work queues.
It follows that the performance storage system for a sequencing analysis cluster does not necessarily have to provide high capacity in addition to high performance. Rather, its capacity only needs to be large enough to store data hotspots, and these working datasets can be staged in and out from slower high-capacity file systems as needed by individual nodes. With the proper automation infrastructure, this approach can significantly reduce the costs associated with procuring high-performance storage tiers by keeping their capacities to a minimum.
Cloud computing resides at the extreme end of this sort of architecture, and eschewing physical clusters in favor of public or private clouds for sequencing analysis has demonstrated some success. A common cloud-based computing architecture uses object storage (such as Amazon S3) to house large volumes of sequencing data and localized block storage for processing; hot data is staged into ephemeral storage when an instance is provisioned and copied back upon completion, foregoing a high-performance storage system altogether.
Applications
Perhaps more than any other area, applications are where the disparity between sequencing-driven computational biology and high-performance computing is most apparent. While traditional HPC applications have been driven to exploit high degrees of parallelism since the advent of massively parallel architectures in the early 1990s, the NGS technologies that have been driving bioinformatics software are much newer. This has resulted in two major factors that frustrate the advancement of the computational aspects of sequence analysis.
Data has not yet standardized
The physics and chemistry underpinning DNA sequencing are still evolving and producing new forms of data. While the As, Ts, Gs, and Cs of sequencing remain the same, a variety of companies are developing new technologies that augment raw sequence reads with metadata that can assist in alignment and variant detection. For each new technology and data type, new algorithms and software have to be developed that often supersede older methods and applications.
Further complicating this is the fact that these advances in sequencing technology are driven by competing commercial interests. As a result, the data types and analysis applications that can utilize these data are highly fragmented across the market, and the computational requirements of DNA sequencing lie in the center of this battlefield. Until these formats eventually converge on some standardized way of expressing this additional metadata, it remains risky for scientific application developers to invest time in optimizing algorithms and code when compatibility may be broken by the release of a new sequencer model.
Software quality remains low
The explosion of bioinformatics led by NGS occurred at a time after the technical barriers to computing had been greatly reduced by highly usable software libraries, tools, and languages. While this has allowed biologists to rapidly develop new computational methodologies using expressive languages like R and Python, these languages are notoriously slow and treat parallelism as second-class citizens. This has left the bioinformatics application space littered with codes that never graduated from proof of concept into production-ready scientific applications.
This lack of attention to scalability and performance is not limited to applications being run at small scale. For example, the Joint Genome Institute (JGI) is a large sequencing operation that sequenced 56 terabases of DNA and burned 20 million core-hours on its internal compute cluster during 2012. Of that massive amount of compute, though, over 95 percent of jobs ran on only a single core. In that same year, JGI consumed an additional 11 million core-hours on Hopper, NERSC’s Cray XE6, with “most using just 24 cores (a single Hopper node) for many instances of a serial code.”[3] By comparison, Trestles (the National Science Foundation’s small jobs cluster) only saw 38 percent of jobs require a single core on in that same year.
This is not to say that there are no bioinformatics applications that contain serious engineering effort. However, even these codes generally sidestep common HPC tools (for example, by implementing parallel for loops in pthreads instead of using OpenMP), which precludes the use of common processor affinity and performance profiling tools. While some the most popular bioinformatics tools are starting to get serious about performance optimization, they often rely on technologies (like Java and its virtual machine environment) that remain at the periphery of HPC.
Future Outlook
There is no arguing that DNA sequencing is rapidly becoming a major consumer of computing cycles alongside the traditional computational science domains. The continuously falling prices of sequencing and the entry of genome-guided diagnostics in the clinical space will guarantee that this trend continues. However, the biggest computational demands of DNA sequencing still reside in the routine processing of raw sequencer output, and the statistical analysis of genomes that drive discoveries in biology and medicine are still currently being done at workstation scales.
As DNA sequencing costs fall and technologies improve, there will be greater volumes of more diverse information to analyze, and this is where complex studies that cannot simply exploit the data-parallelism inherent in genomic data will begin requiring true HPC. When the day comes that bioinformatics becomes limited by computing rather than sequencing, HPC will be ready for it; the limitation will be within the current landscape of single-threaded or shared-memory codes.
Work has already begun in demonstrating proofs of concept to make true high-performance bioinformatics tools, but these optimization projects tend to focus only on subcomponents of the entire alignment and variant calling process. In reality, the end-to-end process of turning raw data into high-quality aligned mappings and called variants should be recognized as a single logical process and optimized as such. The current method of running a dataset through pipelines of discrete applications (each with their own idiosyncrasies) is fundamentally inefficient.
Ideally, bioinformatics applications will follow the same path that traditional computational science domains have and eventually converge on fully integrated, high-performance bioinformatics applications. For example, the LAMMPS molecular dynamics package is a materials simulation code that includes an efficient parallelization foundation, a variety of simulation models on top of that, and the tools to simulate materials under a variety of experimental conditions. There is no reason a high-performance framework for operating on a distributed set of DNA sequence reads cannot be similarly developed, and it is likely that bioinformatics will move in this direction as problems move beyond a single node’s capability.
In a broad sense, these problems that are preventing bioinformatics from truly breaking into HPC are not unique or unexpected; computational physics and chemistry faced many similar challenges in their early days. The difference is that physics and chemistry had the luxury of growing up with HPC whereas DNA sequencing is being thrust into HPC by the unrelenting growth of the DNA sequencing industry. This momentum behind sequencing is now drawing in experts from all fields including HPC, and there is little doubt that the industry will overcome these growing pains in the coming years.
[1] This is not strictly true because Gordon has a second QDR InfiniBand fabric with the same bandwidth reserved for storage.
[2] This is only true for writes on Gordon. Reads from Lustre wind up entering the compute fabric at locality-unaware I/O nodes and typically have to traverse the torus to get to the requesting compute node.
[3] Quoted from High Performance Computing and Storage Requirements for Biological and Environmental Research Target 2017 (DOE Report LBNL-6256E), edited by Richard A. Gerber and Harvey J. Wasserman. Available online http://www.escholarship.org/uc/item/69z9k2wh