

A team from the Joint Genome Institute at Lawrence Berkeley National Lab and researchers from UC Berkeley have used 15,000 cores on the Cray XC30 “Edison” supercomputer to boost the complete assembly of the human genome, bringing the time down to 8.4 minutes.
The work represents how the coupling of high-powered computational capacity, matched with novel approaches to complex code parallelization, can significantly speed large-scale scientific research. For genomics, this speedup marks a dramatic improvement in what was possible before for using de novo assemblers to rebuild a genome from a selection of short reads. For instance, on the same machine, the unmodified Meraculous code took 23.8 hours. For the far more complex wheat genome, full assembly has been difficult for most standard de novo assemblers. The team used their HipMer approach to scale wheat genome assembly across 15,000 cores in just under 40 minutes.
The team says that while the de novo approach has “inherent advantages of discovering variations that may remain undetected when aligning sequence data to a reference genome, unfortunately, de novo assembly computational runtimes cannot up with the data generation of modern sequencers.”
The large-scale parallelization effort, which the team calls HipMer (short for high performance Meraculous—the name of the de novo assembler) picks apart the algorithm for end-to-end parallelization, an effort that involved extensive code work on multiple steps of the genome assembly process. While the full details of the parallelization effort can be found here, the takeaway is that there might be a new approach for genome research centers to get around one of the most pressing computational problems—keeping up with the massive amount of data against extremely complex algorithms.
“For the first time, assembly throughput can exceed the capability of all the world’s sequencers, thus ushering in a new era of genome analysis…The combination of high performance sequencing and efficient de novo assembly is the basis for numerous bioinformatics transformations, including advancement of global food security and a new generation of personalized medicine.”
The Edison supercomputer was designed to tackle this exact type of work—complex scientific codes that are oftentimes just as data-intensive as they are computationally demanding. The Edison supercomputer, which was put into production just over a year ago, was designed to be balanced for both data-heavy jobs and those that need a lot of powerful cores. When the team at NERSC worked with Cray to push out their XC30, the emphasis was on the all-important Cray Aries interconnect, but also on having high memory bandwidth and memory on each node. The end result is a machine with some impressive I/O speeds that match the computational boost from the 124,608 cores. Edison provides 163 gigabytes per second of I/O bandwidth and as a whole, is capable of 2.4 petaflops at peak.
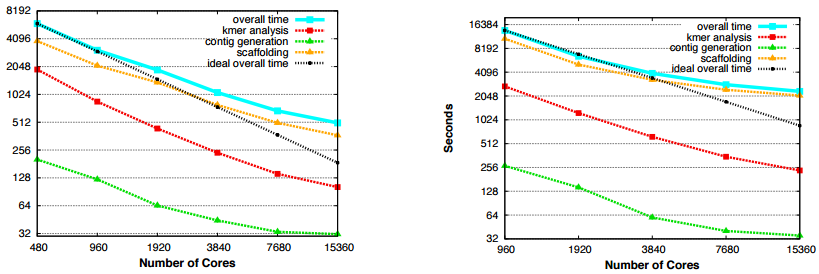
The end-to-end scaling of the team’s HipMer genome assembler approach showing the human genome scalability on Edison on the left and the more complex wheat genome on the right (both axes are in log scale).
Naturally, the supercomputer cores and memory bandwidth are important, but so too is the work put into carving the code so it can efficiently be split over the 15,000 12-core Ivy Bridge cores. The work was built off the ubiquitous Meraculous assembler, which is generally thought to be the most advanced de novo genome assembler.
The resulting HipMer approach is based on “several novel algorithmic advancements by leveraging the efficiency and programmability of UPC, including optimized high-frequency k-mer analysis, communication avoiding de Bruijn graph traversal, advanced I/O optimization, and extensive parallelization across the numerous and complex application phases.” Other improvements include altering th communication model and using a global address space for shared access for reads and writes from any of the many processors.
The team also developed a distributed memory approach for HipMer, which was able to tackle the wheat genome in less than 11 minutes across 20,000 cores. The next phase of the research with HipMer will involve applying it to broader metagenomics studies and extended assembler research.


Holding The Line In The Enterprise
The profit pools in datacenter infrastructure are a bit like oases in the desert: There are a lot of miles between them, and some of them turn out to be mirages. Over the past several decades, the various forms of Hewlett Packard have found and leverage profit pools – it …
Weathering Heights: Of Resolutions And Ensembles
In the past year or so, watching supercomputer maker Cray, which is now part of Hewlett Packard Enterprise, has been a bit like playing a country and western song backwards on the record player. Supercomputing is booming a little (we don’t want to jinx it), Cray has its own interconnect …
NCSA Delta Supercomputer Adopts Slingshot But Forgoes Cray “Shasta” Design
When it comes to supercomputing in academia, the cost of a cluster is almost always an issue and this, coupled with the desire to drive as much compute as possible, drives architectural choices. A great example of this dynamic is the National Center for Supercomputing Applications at the Urbana-Champaign campus …
Be the first to comment